-

-

- GENERAY Chemicals
- GENERAY Markers
- •GENERAY DNA Marker
- •GENERAY Protein Marker
- GENERAY Labwares
-

-

-

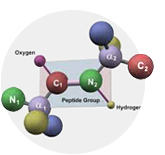
- Peptides Products & Services
- •Custom Peptide Synthesis
- •Peptide Modification
- •Peptide Library Service
- •Peptide Array Service
- •Peptide Synthesis Price
- FAQ
-

-